Pediatría
ISSN impreso:0120-4912
e-ISSN:2444-9369
DOI: 10.14295/rp.v55i1.278
Reporte de caso
Síndrome de sotos: una mirada al gigantismo cerebral. Reporte de caso.
Sotos syndrome: a look at cerebral gigantism. Case report.
Yulys Carolina Redondo Mezaa, Teresa López Garcíab, John Carlos Molina Toroc, Gisel Gordillo Gonzálezd.
a. Pediatra, Universidad del Norte. CEO Mundo Pediátrico CTG. Cartagena, Colombia.
b. Residente de III año, Universidad del Sinú. Cartagena, Colombia.
c. Pediatra, Universidad del Sinú. Cartagena, Colombia.
d. Genetista, profesora Universidad Cooperativa de Colombia. Barranquilla, Colombia.
Recibido 26 de febrero de 2021 Aceptado 06 de julio de 2022
Como Citar: S Redondo Meza YC, López García T, Molina Toro JC, Gordillo González G. Síndrome de sotos: una mirada al gigantismo cerebral. Reporte de caso. Pediatr. 2022;55(1): 46-49.
Autor para correspondencia: Yulys Carolina Redondo Meza.
Correo electrónico: draredondo@gmail.com
Editor Jefe: Fernando Suárez-Obando
Resumen
Antecedentes: El Síndrome de Sotos también conocido como Gigantismo cerebral. Es uno de los síndromes de sobrecrecimiento más frecuentes, La macrocefalia y alta estatura son características frecuente de estos niños. Se caracteriza por una apariencia facial distintiva (frente amplia y prominente con una forma dolicocefalia, escaso cabello frontotemporal, entre otros); discapacidad en el aprendizaje y sobrecrecimiento corporal. El tratamiento va encaminado a favorecer el desarrollo neurológico. Caso clínico: se presenta el caso de un preescolar que en la etapa de lactante evidenció perímetro cefálico aumentado y pobre avance en el neurodesarrollo, con dolicocefalia, frente abombada, fisuras estrechas, columna hipoplásica, narinas apuntando hacia arriba, paladar íntegro, pabellones rotados posteriormente, espalda con cifosis importante lumbar. El estudio molecular, identificó una variante heterocigota, tipo missense c.5165G>C; p.Cys1722Ser en el gen NSD1. El paciente recibe acompañamiento multidisciplinar con avance en neurodesarrollo. Conclusión: A pesar de su distribución mundial, es posible que el síndrome de Sotos no se notifique. Además de su cuadro clínico característico, las pruebas genéticas moleculares también son muy recomendables para llegar al diagnóstico.
Palabras clave: Síndrome de Sotos, gigantismo, megalencefalia.
Abstract
Background: Sotos syndrome is also known as cerebral gigantism. It is one of the most frequent overgrowth syndromes. Macrocephaly and tall stature are typical characteristics of these children. It is characterized by a distinctive facial appearance (broad and prominent forehead with a dolichocephalic shape, sparse frontotemporal hair, among others), learning disabilities, and body overgrowth. Treatment is aimed at promoting neurological development. Clinical case: we present the case of a preschooler who, in the infant stage, showed an increased head circumference and poor progress in neurodevelopment, with dolichocephaly, bulging forehead, narrow fissures, hypoplastic spine, nostrils pointing upwards, intact palate, posteriorly rotated pinnae, and significant lumbar kyphosis. The molecular test identified a heterozygous variant, missense type: c.5165G>C; p.Cys1722Ser in the NSD1 gene. The patient receives multidisciplinary support with progress in neurodevelopment. Conclusion: Despite its worldwide distribution, Sotos syndrome may not be reported. In addition to its characteristic clinical picture, molecular genetic testing is highly recommended for diagnosis.
Key words: Sotos syndrome, gigantism, megalencephaly.
Introducción
El síndrome de Sotos (SS) es un trastorno genético caracterizado por una apariencia facial típica, macrocefalia, crecimiento acelerado, retraso en el desarrollo y una variedad de anomalías asociadas (1,2,3). El sobrecrecimiento característico tiene una incidencia estimada de 1 de cada 15.000 nacimientos. Las variaciones puntuales y deleciones de NSD1, una histona metiltransferasa implicada en la regulación transcripcional, son las responsables de al menos el 75% de los casos de SS (2-6).
Descripción del caso
Se trata de paciente masculino de 3 años y 10 meses, producto de segundo embarazo de madre de 37 años, previamente sana, con alto riesgo obstétrico dado por amenaza de aborto en las primeras semanas. El paciente nace a las 38 semanas por parto vaginal, con antropometrías adecuadas al nacer (Peso al nacer 3620 gr / Talla 52 cm). Hospitalizado a las 48 horas por sospecha de sepsis y meningitis tratada sin secuelas. Como antecedentes personales presenta foramen oval permeable (FOP) y ductus arterioso persistente (DAP) sin repercusión hemodinámica al nacer. Hernia inguinal derecha con incarceración, hidrocele y torsión testicular derecha corregidas a los 3 meses, además de liberación de adherencias que requirió estancia hospitalaria prolongada. Se identifica en los antecedentes familiares que en la línea paterna hay patologías craneales, sin etiología clara.
Durante el desarrollo presento incremento del perímetro cefálico (PC) y pobre avance en el neurodesarrollo, consultó a pediatría en donde la madre comentó, además, erupción dental prematura, sialorrea y respiración bucal.
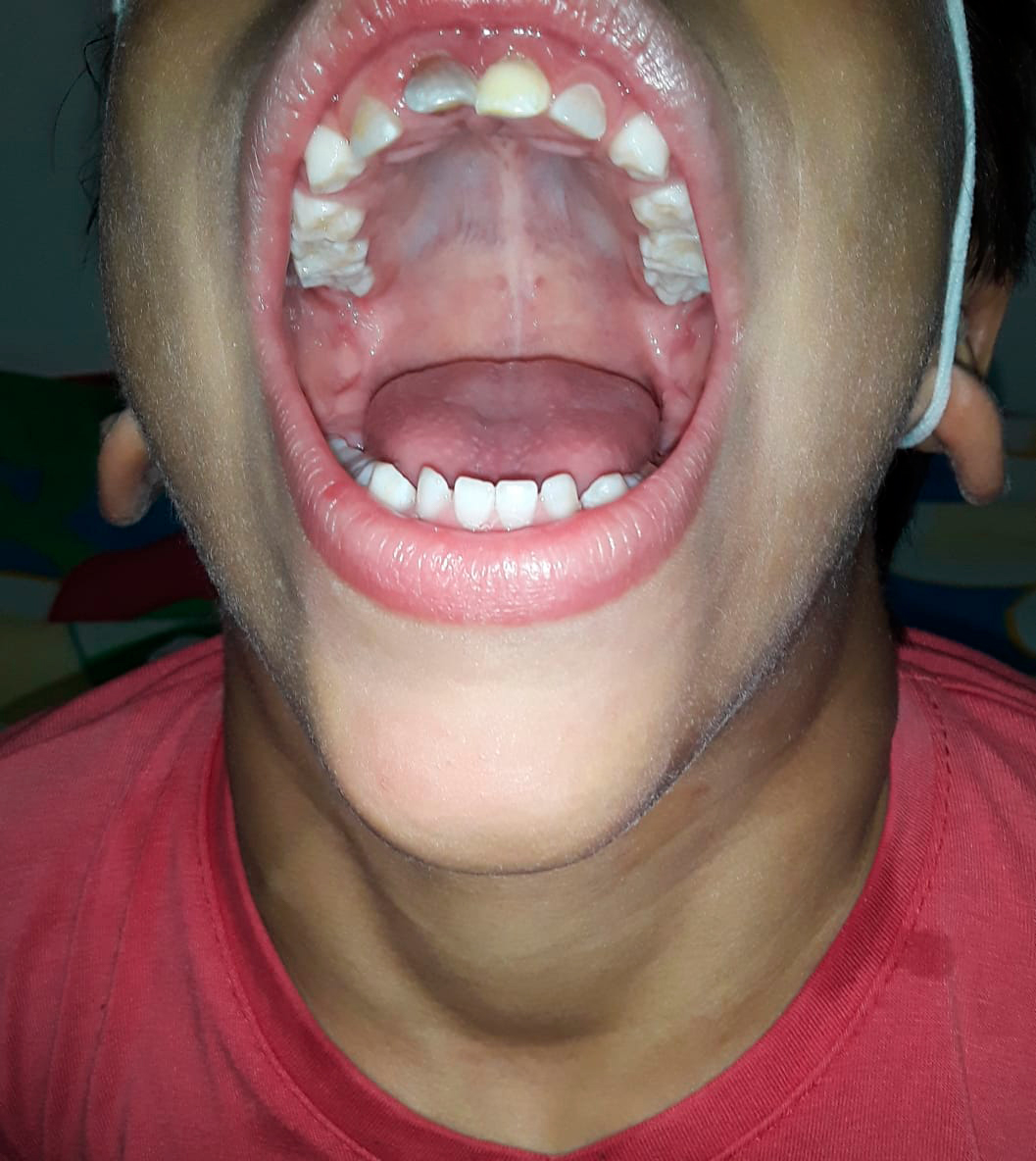
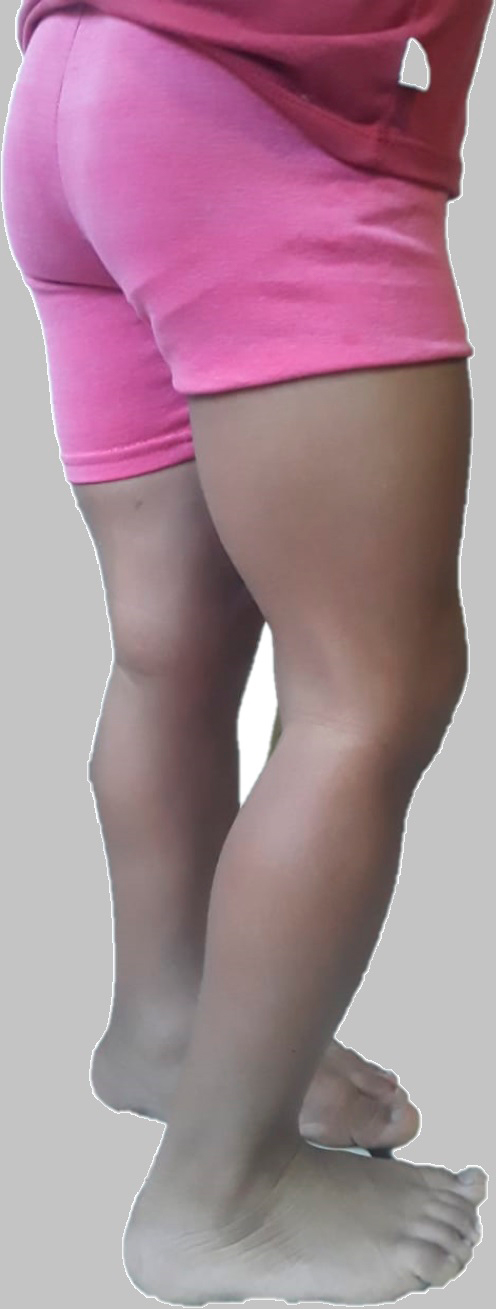
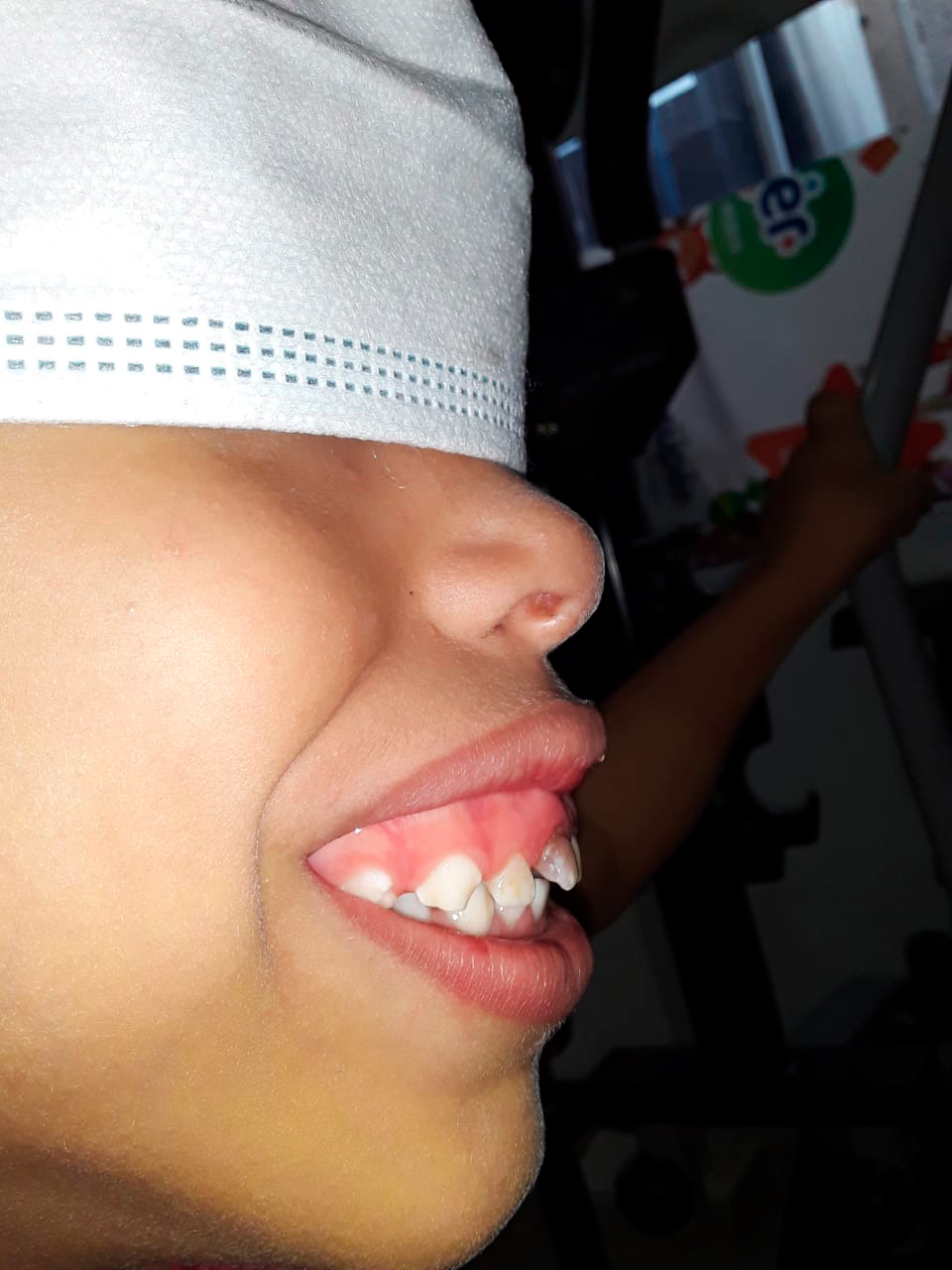
Al examen físico se evidenció, dolicocefalia, frente abombada, fisuras estrechas, columna hipoplásica, narinas apuntando hacia arriba, paladar íntegro, pabellones rotados posteriormente, a la exploración intraoral caries dentales, desgaste oclusal en dentición decidua, mordida abierta anterior y paladar con tendencia ojival (Ver foto 1 y 2), cuello eutrófico, cifosis lumbar, tórax corto, cardiopulmonar normal, abdomen globoso, no masas, hipertrofia de muslo y pie plano bilateral (Ver foto 3).
Entre los paraclínicos realizados se encuentran, potenciales evocados auditivos y visuales normales, resonancia magnética nuclear de cerebro con cavum septum pellucidum como variante anatómica sin evidencia de lesiones. La tomografía axial computarizada (TAC) de cráneo simple con reconstrucción 3D reportó, dolicocefalia y descartó craneosinostosis. Las ecografías cerebral y abdominal total fueron normales. El cariotipo bandeo G, informó un complemento cromosómico 46,XY. Es remitido al servicio de genética quién por los hallazgos al examen ordena estudio molecular, el cual identifica una variante heterocigota, tipo missense c.5165G>C; p.Cys1722Ser en el gen nuclear receptor-binding set domain protein 1 (NSD1). Las variantes patogénicas en este gen son causantes del SS.
Foto 1. Fuente propia
Foto 2. Fuente propia
Foto 3. Fuente propia
Discusión
El síndrome de Sotos, también conocido como gigantismo cerebral o síndrome de deleción cromosómica 5q35, fue descrito por Juan Sotos, por primera vez en 1964. Al contrario que muchas de las alteraciones genéticas del desarrollo, este síndrome puede no ser evidente al nacimiento, necesitando meses o incluso años para ser diagnosticado (2).
A pesar de que su prevalencia es desconocida, es uno de los síndromes de sobrecrecimiento más frecuentes, el segundo en frecuencia luego del síndrome de Beckwith Wiedemann, siendo su incidencia estimada en 1:15 000 nacidos vivos. La macrocefalia y alta estatura es una característica frecuente de estos niños (1).
Se caracteriza por una apariencia facial distintiva (frente amplia y prominente con una forma dolicocefalia, escaso cabello frontotemporal, fisuras palpebrales descendentes, rubor malar, largo y rostro estrecho, mentón largo); discapacidad de aprendizaje (retraso en el desarrollo temprano, intelectual de leve a grave discapacidad); y sobrecrecimiento (altura y / o perímetro cefálico ≥ 2 DE por encima de la media) (8).
Las principales características del SS incluyen problemas de comportamiento (más notablemente trastorno del espectro autista) problemas de adaptación, trastornos de hiperactividad y déficit de atención (7), edad ósea avanzada, anomalías cardíacas, anomalías en cerebrales detectadas a través de resonancia o TAC, hiperlaxitud articular con o sin pie plano (8). El fenotipo completo fue descrito en una revisión sistemática de toda la literatura publicada (1964-2015) que presentó datos empíricos sobre cognición y comportamiento en el síndrome de Sotos (5).
El tratamiento de este cuadro está encaminado a facilitar el correcto desarrollo de las capacidades del niño, con el objetivo de llegar a un desarrollo final dentro de la normalidad. Los programas como estimulación precoz, terapia ocupacional, terapia física, logopedia y educación física adaptativa juegan un papel muy importante en el cuidado de un niño con Síndrome de Sotos (2).
Conclusión
El síndrome de Sotos es una condición autosómica dominante, es un síndrome de sobrecrecimiento, caracterizado por macrocefalia y alta estatura, problemas de comportamiento, edad ósea avanzada, anomalías cardíacas e hiperlaxitud articular. Para el diagnóstico se realiza análisis genético molecular para el gen NSD1 del paciente. El tratamiento va encaminado a favorecer el desarrollo neurológico. Se hace necesario el diagnóstico oportuno para el inicio del manejo y mejorar la calidad de vida del paciente.
Referencias
1. Caino, D., Moresco, A., Breitman, F., Fano, V. Crecimiento en niños con síndrome de Sotos. Medicina Infantil. 2013;20(2):117- 121.
2. DISCAPNET. (15 de Feb de 2018). DISCAPNET. Disponible en: El portal de las personas con discapacidad. https://www.discapnet.es/sindrome-de-sotos.
3. Faravelli, F. Mutaciones NSD1 en el síndrome de Sotos. Am J Med Genet C Semin Med Genet. 2005;137(1):24-31.
4. Kamal, N., Althobiti, J., Alsaedi, A., Bakkar, A., Alkaabi, T. Síndrome de Sotos. Medicina. 2018; 97(47).
5. Lane, C., Milne, E., Freeth, M. Cognition and Behaviour in Sotos Syndrome. Plos One, 2016;11(2): e0149189.
6. Lapunzina, P. Síndrome de Sotos. Protoc diagn ter pediatr, 2010;(1):71-79.
7. Madi, M., Babu, S., Shetty, S., Madiyal, A., Achalli, S., Bhat, S. Síndrome de Sotos. Applied Medical Research. 2016;2(3):63-67.
8. Tatton-Brown, K., Cole, T., & Rahman, N. (2019). Sotos Syndrome. GeneReviews. Obtenido de: https://www.ncbi.nlm.nih.gov/books/NBK1479/.