Pediatría
ISSN impreso:0120-4912
e-ISSN:2444-9369
DOI: 10.14295/rp.v55i2.312
Revisión
Enfermedad de Hirschsprung, un enfoque practico
Hirschprung`s disease, a practical approach
Nicolás Zuluaga Arbeláeza, Santiago Posada Lópezb.
a. Residente de 3er año de Medicina interna Universidad CES. Medellín, Colombia.
b. Medico general Universidad CES. Medellín, Colombia.
Recibido 21 de septiembre de 2021 Aceptado 18 de mayo de 2022
Como Citar: Zuluaga Arbeláez N, Posada López S. Enfermedad de Hirschsprung, un enfoque practico. Pediatr. 2022;55(2):91-97.
Autor para correspondencia: Santiago Posada López
Correo electrónico: santi0117@outlook.com
Editor adjunto: Alvaro León Jácome Orozco
Resumen
La enfermedad de Hirschsprung fue descrita por el pediatra danés Harald Hirschsprung en 1888. Es la neuropatía entérica congénita más común, donde se produce una ausencia de relajación del musculo entérico, con posterior obstrucción intestinal. Su etiopatogenia está dada por la aganglionosis en el tracto digestivo, afectando principalmente la región rectosigmoidea. Así, manteniendo contraído el esfínter anal interno. Se clasifica en cuatro tipos: segmento corto, la más común; segmento largo, segmento ultracorto y aganglionosis coli. La clínica frecuentemente inicia en el periodo neonatal. La triada clásica consiste en distensión abdominal, ausencia de meconio en las primeras 24 a 48 horas de vida y vomito bilioso con intolerancia a la vía oral. La aproximación diagnostica se realiza mediante rayos X de abdomen, enema de contraste o manometría rectal. La prueba gold standard es la biopsia rectal, la cual se considera positiva si hay ausencia de células ganglionares en el plexo mientérico y submucoso del colon. El manejo inicial de la enfermedad se basa en resucitación con líquidos endovenosos, antibiótico de amplio espectro para prevención de enterocolitis y translocación bacteriana, descompresión con sonda nasogástrica y lavados colónicos. El tratamiento definitivo es la resección colónica del segmento afectado. Las principales complicaciones postoperatorios son: excoriación perianal, constipación, suciedad, diarrea, incontinencia fecal y enterocolitis.
Palabras clave: Enfermedad de Hirschsprung, manejo, Megacolon Agangliónico Congénito, diagnóstico.
Abstract
Hirschsprung's disease was described by the Danish pediatrician Harald Hirschsprung in 1888. It is the most common congenital enteric neuropathy, with a lack of relaxation of the enteric muscle, with subsequent intestinal obstruction. Its etiopathogenesis is given by aganglionosis in the digestive tract, mainly affecting the rectosigmoid region. Thus, keeping the internal anal sphincter contracted. It is classified into four types: short segment, the most common; long segment, ultrashort segment, and aganglionosis coli. The clinical presentation frequently begins in the neonatal period. The classic triad consists of abdominal distention, absence of meconium in the first 24 to 48 hours of life, and bilious vomiting with intolerance to the oral route. The diagnostic approach is performed by abdominal X-rays, contrast enema, or rectal manometry. The gold standard test is the rectal biopsy, which is considered positive if there are no ganglion cells in the myenteric and submucosal plexus of the colon. The initial management of the disease is based on intravenous fluid resuscitation, broad-spectrum antibiotics to prevent enterocolitis and bacterial translocation, decompression with a nasogastric tube, and colonic lavage. The definitive treatment is colonic resection of the affected segment. The main postoperative complications are perianal excoriation, constipation, dirt, diarrhea, fecal incontinence, and enterocolitis.
Key words: Hirschsprung's Disease, management, Congenital Aganglionic Megacolon, diagnosis.
Introducción
La ausencia del paso del meconio en un neonato a término obliga descartar la presentación de una enfermedad causada por la ausencia de células ganglionares en los plexos mientéricos del intestino distal; o también conocido como enfermedad de Hirschsprung (1,2).
Cirujanos hindúes describieron una enfermedad análoga llamada «Baddha Gudodaram» y posteriormente toma su nombre del médico Harald Hirschprung, pediatra danés, quien describió por primera vez en 1888 el caso de dos niños con constipación y distensión abdominal que luego fallecieron, encontrando en sus necropsias gran dilatación e hipertrofia del colon transverso y descendente, sin una causa clara (3).
Solo hasta 1901 se pudo reconocer la ausencia de células ganglionares en el plexo de Auerbach y de Meissner en el segmento intestinal distal no dilatado como la causa de la obstrucción intestinal (4).
es la neuropatía entérica congénita más común, donde la ausencia de relajación del musculo entérico lleva a una obstrucción intestinal funcional, siendo la causa más frecuente en neonatos, y menos común en preescolares y escolares (5). Afecta aproximadamente a 1 de cada 5000 recién nacidos, más en varones, con una relación de 4:1 con respecto a las mujeres (6).
Se caracteriza por tener un patrón hereditario multifactorial. Se presenta de manera aislada hasta en el 70 % de los casos, pero hay alta asociación con otros síndromes y cromosomopatías (Ver Tabla 1) (1,2).
Etiología
El tracto gastrointestinal requiere, para lograr un movimiento peristáltico funcional, un flujo sanguíneo y metabólico adecuado y que el funcionamiento del sistema nervioso entérico (SNE) sea optimo (7).
La cresta neural, deriva del tubo neural y se forma durante el periodo embrionario. Sus células se separan y localizan a ambos lados del tubo neural, migrando lejos de su origen en dirección cráneo caudal, para formar los ganglios de la raíz posterior, componentes del sistema nervioso autónomo incluyendo los plexos mientéricos del intestino y los nervios craneales V, VII, IX y X (8).
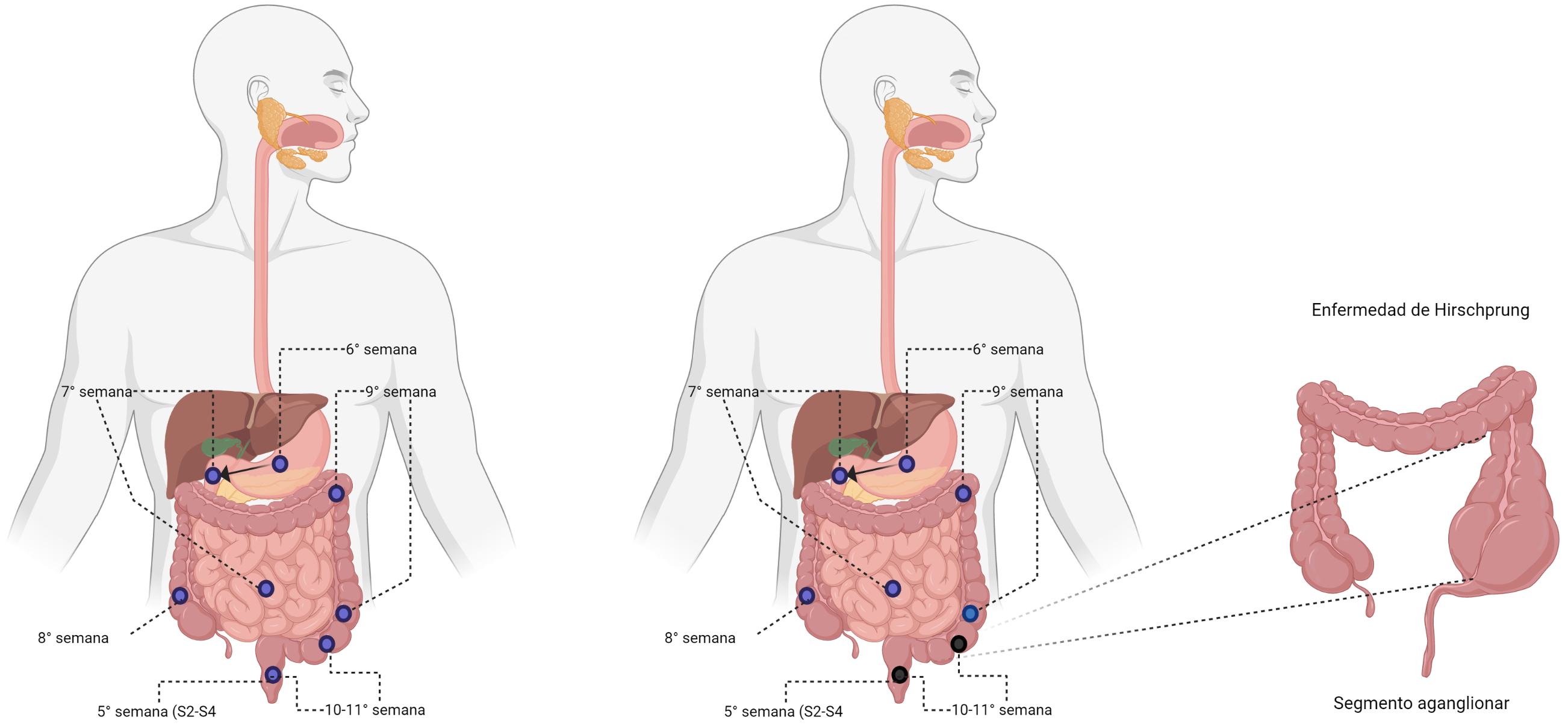
Bajo condiciones normales, las células de la cresta neural migran en dirección céfalo-caudal, entre la 5ª y 12ª semana de gestación, desde el área vagal de la cresta neural a lo largo de los nervios vagales hasta el mesénquima del intestino anterior, luego se extienden por todo el intestino, (de manera progresiva y sin interrupción) inervando primero el intestino proximal y por último el recto (9). La interrupción de este proceso, conducen a disganglionosis del intestino de manera longitudinal, explicando, porque todos los casos de la aganglionosis involucran principalmente la región rectosigmoide(10).
En la aganglionosis solo está documentada la falla en la migración, en caso de fallo en proliferación, supervivencia o diferenciación se da la disganglinosis que puede ser por pocas células ganglionares o mala función de estas (10).
Sin células ganglionares, los segmentos afectados permanecen contraídos, por efecto simpático permanente y ausencia de relajación por falta de estímulo parasimpático. El esfínter anal interno no puede relajarse ante la presencia de materia fecal, conduciendo a una falta de motilidad intestinal, estasis fecal y estreñimiento (9).
Clasificación
Puede ser de 4 tipos: de segmento corto, donde solo se afecta la región rectosigmoidea del intestino, la cual constituye el 80% de los pacientes; de segmento largo, definido como aganglionosis más allá de la flexión sigmoidea, la cual representa el 10 % de los casos; de segmento ultracorto, el 5 % de los afectados y aganglionosis coli con o sin compromiso del íleon distal, en el 5 % restante (5). En casos más raros puede afectarse también la parte distal del intestino delgado, donde todavía no está claro si corresponde a una forma extensa de enfermedad de Hirschprung o a una expresión diferente de la enfermedad (9,11).
Otro concepto discutido, es la presencia de enfermedad parcheada o skip lesion donde las regiones aganglionares se encuentran fragmentadas en distintas zonas, lo cual puede llevar a errores de diagnóstico y de tratamiento (5)2
Existe una entidad relacionada, llamada acalasia del esfínter anal interno, previamente denominada segmento ultracorto, donde el segmento agangliónico es tan pequeño que resulta muy difícil hacer un diagnóstico mediante biopsia rectal, y requiere complementar con manometría anorrectal para evidenciar la presencia del reflejo recto-anal inhibitorio, el cual está presente cuando hay una relajación mayor al 25 % de la presión basal del canal anal (12,13,31).
Genética
La enfermedad de Hirschsprung en el 12 % de los casos se asocia a otros síndromes genéticos, siendo el síndrome de Down la entidad más común, presentando un riesgo 100 veces mayor de padecer la enfermedad, que el resto de la población (14,15).
Hasta el 20% de los casos son familiares, con herencia autosómica dominante con penetrancia variable y están implicados varios factores de susceptibilidad. Se han identificado más de diez genes involucrados en la migración de las células de la cresta neural (3).
Las mutaciones más comunes ocurren en el protooncogen RET, 10q11.2, del inglés: Rearranged during transfection protooncogene, que codifica para un receptor de membrana con actividad tirosincinasa expresado en tejidos y tumores derivados de la cresta neural, el cual se relaciona con neoplasia endocrina múltiple (MEN2B, del inglés: Multiple endocrine neoplasia, type 2B) (14). Otro gen importante es el gen que codifica el receptor de endotelina B (ENDRB), 13q22.3, relacionado con el síndrome de Shah-Waardenburg (WS4); y el gen ZEB2, 2q22.3 (Del inglés:Zinc finger e box-binding homeobox 2), involucrado en la migración de células de la cresta neural (figura 1) y desarrollo de estructuras situadas en la línea media. El gen ZEB2 está relacionado con el síndrome de Mowat-Wilson (MWS) en el cual, la enfermedad de Hirschprung se presenta hasta en el 43.4 % de los casos (7,16,17).
Estudios más recientes han evidenciado variantes patogénicas en el gen que codifica la actina gama 2 (ACTG2, 2p13.1), el cual se expresa en el músculo liso entérico y de la vejiga, donde se altera la motilidad intestinal y ocurren síntomas obstructivos funcionales siendo esta otra causa probable de la enfermedad de Hirschsprung (18).
Las malformaciones asociadas ocurren hasta en el 35 % de los casos, que pueden incluir microcefalia, dimorfismo facial, agenesia del cuerpo calloso, y anomalías de los miembros, haciendo parte de las neurocistopatías (3,19).
Presentación clínica
Se espera dismotilidad intestinal de forma secundaria al colon aganglionar, debido a una contracción sostenida del segmento denervado, con incapacidad para la relajación y el paso impedido de las heces, se instaura así, un cuadro de obstrucción intestinal funcional característico (10,18).
Este cuadro sintomático inicia frecuentemente en el periodo neonatal, dado por una triada clásica de distensión abdominal, ausencia de paso del meconio en las primeras 24 a 48 horas de vida y la presencia de vómito bilioso con intolerancia a la vía oral (20). Sin embargo, en ocasiones, la enfermedad que afecta segmentos cortos expresa signos y síntomas menos alarmantes como heces encintadas y dispepsia, que pueden pasar por alto la enfermedad y evitar el diagnóstico temprano. De forma poco habitual, en niños mayores, puede encontrarse la enfermedad, con el antecedente de constipación crónica, severa, de inicio temprano y mala respuesta al tratamiento. Incluso, menos del 1 % de los pacientes son diagnosticados en la adultez, y su única manifestación es la constipación (21).
Por otro lado, hasta un tercio de los pacientes pueden debutar con una entidad conocida como enterocolitis asociada a la enfermedad de Hirschsprung (EAEH). Esta es la complicación más grave y potencialmente fatal de la enfermedad, con morbilidad y mortalidad significativa, caracterizada por fiebre, diarrea explosiva, distensión abdominal, vómitos, salida de flatos explosivos y heces líquidas durante el examen rectal (22). Ocurre por estasis fecal que conduce a proliferación bacteriana excesiva e inflamación de la mucosa, además pueden contribuir a su desarrollo, la inmunidad anormal de la mucosa intestinal y la disbiosis de la flora intestinal. Si no se trata, puede provocar perforación intestinal, shock séptico e incluso la muerte. En general, 18 % a 50 % de los pacientes con enfermedad de Hirschprung pueden desarrollar enterocolitis a lo largo de su vida (9).
El manejo clave incluye la reanimación temprana y antibióticos de amplio espectro y descompresión intestinal. Es potencialmente mortal hasta en el 60 % de los pacientes, por lo que no debe subestimarse y debe tratarse de forma oportuna (22).
Es importante evaluar características dismórficas asociadas, en particular el fenotipo de síndrome de Down, anomalías espinales, adecuada implantación del ano y otros signos de neurocristopatía como el mechón de pelo blanco o nevus cutáneos múltiples (13).
Tabla 1. Síndromes asociados a enfermedad de Hirschsprung
|
Síndrome de Down |
|
Síndrome de Shah-Waardenburg |
|
Síndrome de Smith-Lemli-Optiz |
|
Síndrome de Haddad |
|
Síndrome de Mowat-Wilson |
|
Síndrome de hipoventilación congénita |
|
Neoplasia Endocrina Múltiple (MEN) tipo IIA y IIB |
|
Cromosoma X frágil |
Figura 1. Migración de células de la cresta neural.
Enfermedad de Hirschsprung. A la izquierda migración craneocaudal completa de las células de la cresta neural, a lo largo del todo el tracto gastrointestinal. En el centro, migración craneocaudal incompleta de las células de la cresta neural, dejando un segmento aganglionar, que origina la enfermedad. Desarrollado con BioRender
Diagnóstico
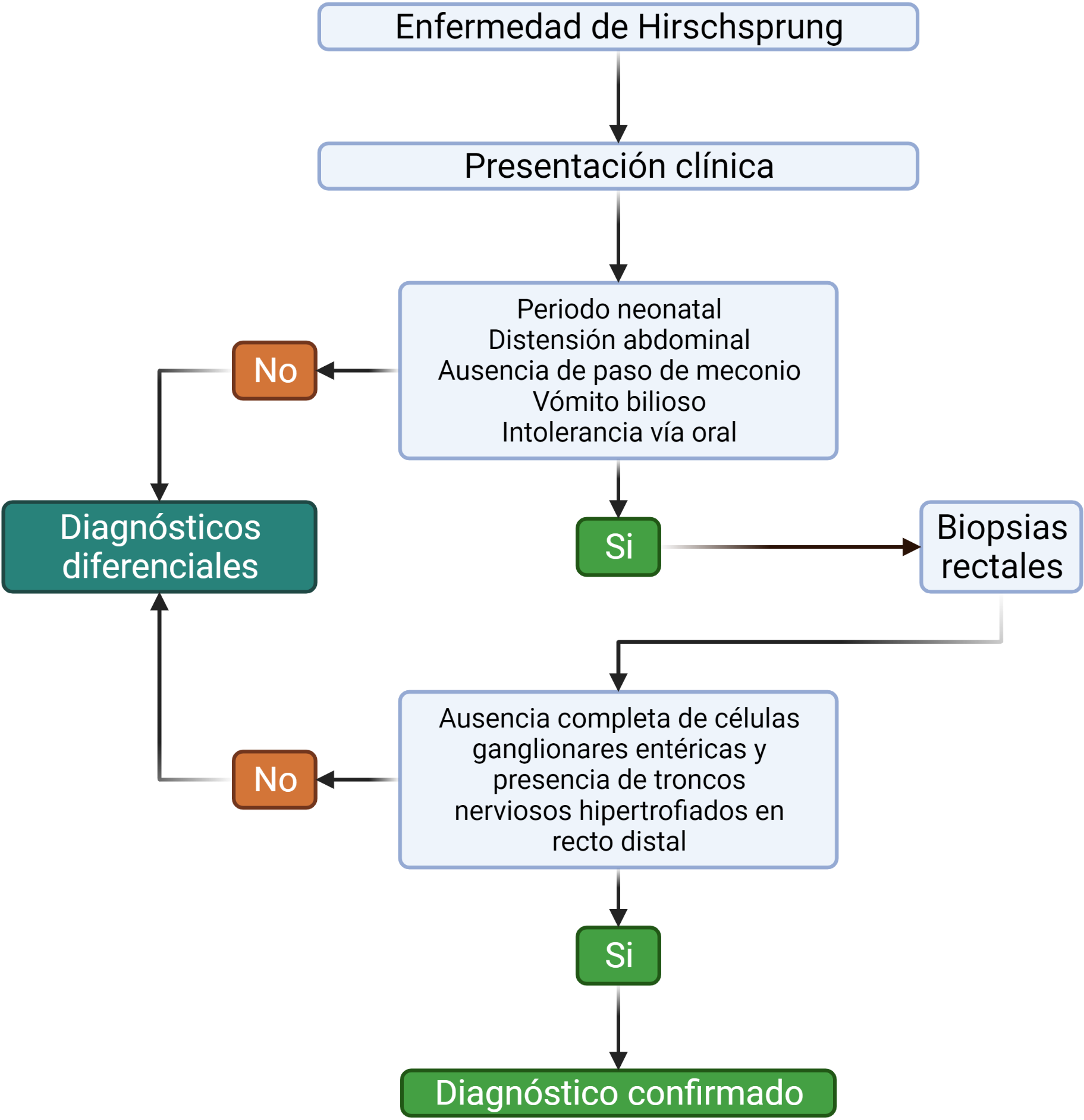
La aproximación inicial se hace mediante una radiografía de abdomen simple donde se encuentran asas intestinales dilatadas, ausencia de heces y gases en el recto, hallazgos inespecíficos pero que ayudan a descartar otras etiologías (9).
Mediante un enema de contraste se puede hacer un mapa topográfico de la anatomía del colon, evidenciando en la EH, una parte del colon dilatado y un segmento distal estrecho, la unión de estos dos segmentos es conocida como zona de transición y tiene forma de cono. La porción dilatada corresponde al colon normal y el área estrecha es la porción aganglionar, permanentemente contraída(9).
El enema contrastado tiene una sensibilidad del 86 % y especificidad del 90 %, con falsos negativos de hasta 20 al 28 %, por lo que si se encuentra alterado siempre es necesario ampliar los estudios. Si se sospecha enterocolitis no debe realizarse en el curso de esta por riesgo de perforación intestinal (23).
Otra ayuda diagnóstica es la manometría rectal que mide la presión y el reflejo inhibitorio del músculo del esfínter anal interno, el cual está ausente en la EH con un valor predictivo negativo del 100 %. Puede realizarse en recién nacidos pretérmino de hasta 26 semanas (9).
Si bien la presencia de zona de transición en el enema con contraste sugiere el diagnostico, siempre se requiere demostrar la ausencia completa de células ganglionares entéricas y la presencia de troncos nerviosos hipertrofiados en el plexo submucoso y mientérico a nivel del recto distal por medio de la biopsia rectal (18, 24).
Para llevarla a cabo, se debe tomar de dos a tres muestras de la pared posterior del recto a 1, 2 y 3 cm por encima de la línea dentada, respectivamente, ya que la población de células ganglionares disminuye progresivamente en el recto distal. Debe ser de al menos 3 mm de diámetro y la submucosa debe tener al menos un tercio del grosor de la muestra para representar la muscular de la mucosa (9). La biopsia convencional, que se realiza en el país, se obtiene bajo visión directa, no se requiere descompresión del colon y se toma muestra de la muscular o de espesor completo, tiene mayor riesgo, pero mejor rendimiento diagnostico (10, 21). Las complicaciones asociadas a la biopsia rectal son poco frecuentes pero incluyen sangrado, cicatrices, estenosis, perforación, sepsis y riesgos de la anestesia (18).
Las pruebas de inmunohistoquímica mejoran la precisión mediante la aplicación de acetilcolinesterasa, que muestra fibras nerviosas engrosadas de más de 40 μm e inmunohistoquímica de calretinina que evidencia ausencia de inmunorreactividad de calretinina. Expresiones que son patognomónicas de este fenómeno, reportando así el diagnóstico definitivo por un patólogo experto. La calretinina es la más usada, pero puede también ordenarse enolasa neuronal especifica y proteína S100 (22).
Es importante considerar la inmadurez del sistema nervioso entérico de los bebés prematuros, en los cuales las muestras pudiesen ser erróneas. Además, este grupo poblacional es más susceptible de sufrir perforación o sepsis luego del procedimiento. Se recomienda esperar hasta la semana 37 de edad gestacional corregida antes de realizar la intervención (24).
Hallazgos histológicos
En la biopsia rectal el hallazgo diagnóstico es la ausencia de células ganglionares dentro de los plexos mientérico y submucoso del colon, la presencia de una sola célula ganglionar excluye el diagnóstico de EH clásica. Más del 90% de los pacientes también tienen fibras nerviosas extrínsecas hipertróficas (9).
Diagnóstico diferencial
Los principales diagnósticos diferenciales se aprecian en la tabla 2.
Tratamiento
El manejo incluye, la resucitación con bolos de líquidos endovenosos en caso de signos de deshidratación o shock hipovolémico, pueden ser necesarios varios bolos. Se debe iniciar cuanto antes un antibiótico intravenoso de amplio cubrimiento para prevenir la enterocolitis y la translocación bacteriana (3).
Se debe realizar descompresión gástrica con sonda nasogástrica y colónica mediante lavados con 10 ml / kg de solución salina tibia, lo cual que puede generar la salida explosiva de heces. Deben realizarse varias veces por día según sea necesario y es fundamental para prevenir la enterocolitis y antes de la cirugía. Las dilataciones anales son un complemento útil para el lavado rectal (3). La sonda rectal se debe introducir más allá de la zona aganglionar, el paso de líquido debe ser a baja presión y la obtención del enema debe realizarse sin retirar la sonda para ayudar el paso a través de la zona afectada.
Luego de confirmar el diagnóstico, la cirugía es el tratamiento definitivo, lo primero es realizar toma de biopsias, seguido de un examen patológico, para saber cuánto intestino está comprometido y necesitará ser resecado, luego se remueve el segmento agangliónico, y posteriormente se realiza anastomosis del segmento proximal normal con el recto distal o canal anal (9) (24).
Existen diferentes abordajes quirúrgicos los cuales pueden realizarse en uno o dos tiempos operatorios. La técnica estará supeditada a la presencia de otras malformaciones digestivas, edad y necesidad de colostomía derivativa previa. Sin embargo, la tendencia actual es la realización de la cirugía a la edad más temprana posible y en un solo tiempo quirúrgico (26).
Clásicamente se han descrito cuatro procedimientos: Swenson (abordaje transanal, rectosigmoidectomía convencional o laparoscópica con resección y anastomosis termino-terminal realizada mediante la esterilización del intestino a través del ano); Duhamel (descenso retrorrectal del sector colónico sano, deja el recto en su sitio y desplaza el intestino con células ganglionares hacia el espacio retrorrectal); Soave (abordaje abdominal convencional y transanal en 2 tiempos, disección endorrectal y ablación de la mucosa del segmento agangliónico distal llevando el intestino con células ganglionares hacia el ano) y Soave Boley (procedimiento en un único acto quirúrgico) y el también conocido procedimiento de La Torre (3,5,27).
La técnica de pull-throug’ transanal o De la Torre Mondragón es el resultado de los múltiples avances en el diagnóstico y manejo poco invasivo de la enfermedad de Hirschsprung. Se realiza un descenso del sector agangliónico transanal sin ingresar a la cavidad abdominal (5).
En presencia de segmento ultracorto de la enfermedad de Hirschsprung, con compromiso limitado al esfínter anal interno también llamado acalasia, se corrige mediante esfinterotomía. Por el contrario, cuando es de segmento largo, y está comprometido todo el colon, y a veces parte del íleon, se debe hacer una anastomosis íleo-anal (28).
La cirugía es efectiva a corto plazo, pero pueden persistir problemas funcionales y complicaciones asociadas incluyendo disfunción intestinal, constipación, incontinencia o suciedad fecal y enterocolitis (22). Se recomienda que los cuidadores se entrenen en la técnica de lavado colónico, con el fin de continuar su uso en casa por programación o a necesidad según los hábitos fecales en el post operatorio.
La enterocolitis asociada a EH sigue siendo una causa de morbi-mortalidad importante, puede ocurrir en cualquier momento durante el curso de la enfermedad. La incidencia reportada antes de la cirugía oscila entre 6 y 50 %, y después de la cirugía del 2 al 35 % (22, 29).
Otras alternativas terapéuticas
Una alternativa nueva y novedosa consiste en la terapia de reemplazo celular, con la producción exitosa de neuronas entéricas funcionales a partir de células madre pluripotenciales humanas inducidas, lo cual ha aumentado la esperanza de la medicina regenerativa como una opción de tratamiento para pacientes con EH. Estas células también se han utilizado para corregir las mutaciones genéticas asociadas mediante la edición del genoma (22).
Tabla 2. Diagnósticos diferenciales de la enfermedad de Hirschsprung.
|
Causas estructurales: |
Atresia, malformación ano rectal, estenosis, malrotación, vólvulo. |
|
Causas relacionadas con la motilidad: síndrome |
Síndrome de colon izquierdo pequeño |
|
Causas de bloqueo |
Tapón de meconio, íleo de meconio por fibrosis quística |
|
Otras: |
Hipotiroidismo, hipercalcemia, prematuridad, sepsis, constipación idiopática. |
|
Causas maternas: |
Infecciones, diabetes, uso de medicamentos o drogas ilícitas |
Complicaciones
Las complicaciones postoperatorias pueden ser tempranas o tardías. Incluyen excoriación perianal, constipación, suciedad, diarrea, incontinencia fecal y enterocolitis. Los dos primeros a menudo se resuelven en pocos meses y el tratamiento es sintomático (13).
Las estenosis pueden ser por fugas de la anastomosis o estenosis rectal y generalmente se tratan mediante dilatación anal diaria o semanal. La acalasia del esfínter anal puede tratarse con inyecciones de Botox, aplicaciones tópicas de óxido nitroso o rectomiomectomía posterior (30).
La suciedad e incontinencia fecal pueden ocurrir por lesión del esfínter anal durante la cirugía. La etiología de la constipación es variada, desde estenosis, aganglionosis residual o recurrente, hasta acalasia del esfínter anal. Se maneja aumentando la fibra en la dieta y administrado antidiarreicos o laxantes, según la condición. Todos estos síntomas afectan negativamente no solo la función gastrointestinal sino también la calidad de vida tiene repercusiones psicológicas, conductuales y del desarrollo. El 60 % de los pacientes experimenta complicaciones a largo plazo, y el 10 % necesita alguna cirugía adicional o una colostomía después del tratamiento inicial (3,30,24).
Aspectos claves de la enfermedad
La EH es la causa más común de obstrucción intestinal baja en neonatos, tiene un patrón hereditario multifactorial y se define por la ausencia de células ganglionares en los plexos mientéricos y submucosos del intestino distal.
El cuadro sintomático se caracteriza por, distensión abdominal, ausencia del paso del meconio, vómito bilioso e intolerancia a la vía oral. El gold standard diagnóstico es la biopsia rectal. El tratamiento es quirúrgico. Ver figura 2 en donde se expone el flujograma diagnóstico.
Figura 2. Algoritmo diagnostico/cuadro sinóptico
Referencias / Bibliografía
1. Schäppi MG, Staiano A, Milla PJ, Smith VV, Dias JA, Heuschkel R, et al. A Practical Guide for the Diagnosis of Primary Enteric Nervous System Disorders: J Pediatr Gastroenterol Nutr. noviembre de 2013;57(5):677-86.
2. Tabbers MM, DiLorenzo C, Berger MY, Faure C, Langendam MW, Nurko S, et al. Evaluation and treatment of functional constipation in infants and children: evidence-based recommendations from ESPGHAN and NASPGHAN. J Pediatr Gastroenterol Nutr. 2014;58(2):258–74.
3. Moore SW. Advances in understanding functional variations in the Hirschsprung disease spectrum (variant Hirschsprung disease). Pediatr Surg Int. 2017;33(3):285-98.
4. Torre-Mondragón L. Enfermedad de Hirschsprung. Mitos y realidades a 120 años de su descripción. Acta Pediatr Mex. 2008;29(3):139-146.
5. Carro Gabriela, Ormaechea Martín, Silva Ema Da, Juambeltz Carlos. Enfermedad de Hirschsprung: resultados del tratamiento quirúrgico en el Hospital Pediátrico Pereira Rossell. Arch. Pediatr. Urug. 89( 3 ): 158-164.
6. Sajjad N, Hilal K, Khandwala K, Arshad M, Uddin N. Usefulness of delayed films of contrast enema for detecting Hirschsprung’s disease. Cureus. 2019;11(12):e6339.
7. Butler Tjaden NE, Trainor PA. The developmental etiology and pathogenesis of Hirschsprung disease. Transl Res. 2013;162(1):1- 15.
8. Amiel J, Lyonnet S. Hirschsprung disease, associated syndromes, and genetics: a review. J Med Genet. 2001;38(11):729– 39.
9. Kroll-Wheeler L, Wilson AM. Educational Case: Hirschsprung Disease. Acad Pathol. 1 de enero de 2019;6:237428951989308.
10. Hackam D, Grikscheit T, Wang K, Upperman J, Ford H. Chapter 39: Pediatric Surgery. In: Andersen D, Dunn D, Hunter J, ed. by. Schwartz’s principles of surgery 11th ed. Mc Gram Hill; 2019.
11. Moore SW. Total colonic aganglionosis in Hirschsprung disease. Semin Pediatr Surg. 2012;21(4):302-9.
12. Raveenthiran V. Knowledge of ancient Hindu surgeons on Hirschsprung disease: evidence from Sushruta Samhita of circa 1200-600 bc. J Pediatr Surg. 2011;46(11):2204-8.
13. Das K, Mohanty S. Hirschsprung Disease — Current Diagnosis and Management. Indian J Pediatr. 2017;84(8):618-23.
14. Mc Laughlin D, Puri P. Familial hirschsprung’s disease: a systematic review. Pediatr Surg Int. 2015;31(8):695-700.
15. Moore SW. Advances in understanding the association between Down syndrome and Hirschsprung disease (DS–HSCR). Pediatr Surg Int. 2018;34(11):1127-37.
16. Villota D VA, Saldarriaga G W, Gómez C JF. Síndrome de Mowat- Wilson: caso clínico. Rev Chil Pediatría. 2012;83(4):371-6.
17. Coyle D, Puri P. Hirschsprung’s disease in children with Mowat– Wilson syndrome. Pediatr Surg Int. 2015;31(8):711-7.
18. Moore SW, Maluleke T, El Hosny AA. Is Hirschsprung disease a purely neurological condition? A study of the Actin G2 smooth muscle gene in Hirschsprung disease. J Pediatr Surg. 2019;54(10):2028-31.
19. Jiménez, J. and Fernandez, L., n.d. Enfermedad de Hirschsprung. AEPED, pp.47-52.
20. Wetherill C, Sutcliffe J. Hirschsprung disease and anorectal malformation. Early Hum Dev. 2014;90(12):927-32.
21. Muise ED, Cowles RA. Rectal biopsy for Hirschsprung’s disease: a review of techniques, pathology, and complications. World J Pediatr. 2016;12(2):135-41.
22. Tam PKH, Chung PHY, St Peter SD, Gayer CP, Ford HR, Tam GCH, et al. Advances in paediatric gastroenterology. The Lancet. 2017;390(10099):1072-82.
23. Cathia Selman; Constanza Alzola Internas 7° año, Facultad de Medicina, Universidad de Chile. Enfermedad de Hirschprung: Avances en el diagnóstico. Rev. Ped. Elec. 2016;13(2):13-18.
24. Westfal ML, Goldstein AM. Pediatric enteric neuropathies: diagnosis and current management. Curr Opin Pediatr. 2017;29(3):347-53.
25. Massardo V Teresa, Jaimovich F Rodrigo, Rodríguez T J. Carlos, Azolas A Cristián, Saavedra S José, Lillo G Ricardo et al . Tromboembolismo pulmonar en lactante menor, portadora de enfermedad de Hirschsprung: caso clínico. Rev. chil. radiol. 2005;11(1): 32-35.
26. Alberto Peña. Enfermedad de Hirschsprung Los avances y las preguntas no contestadas. Cir. Pediatr. 2002; 15: 46-47.
27. Suita S, Taguchi T, Ieiri S, Nakatsuji T. Hirschsprung’s disease in Japan: analysis of 3852 patients based on a nationwide survey in 30 years. J Pediatr Surg. enero de 2005;40(1):197-202.
28. PÉREZ BILLI LUIS E, BENEDICTTI JUAN L, GUTIÉRREZ CARMEN, GALIANA HORACIO GUTIÉRREZ. Enfermedad de Hirschsprung con afectación total del colon: primer caso nacional con la técnica quirúrgica de Lester Martin modificada. Arch. Pediatr. Urug. 2001;72(1): 34-37.
29. Nakamura H, Lim T, Puri P. Inflammatory bowel disease in patients with Hirschsprung’s disease: a systematic review and meta-analysis. Pediatr Surg Int. 2018;34(2):149-54.
30. Green HL, Rizzolo D, Austin M. Surgical management for Hirschsprung disease: A review for primary care providers. J Am Acad Physician Assist. 2016;29(4):249.
31. Ciriza-de-los-Ríos, Constanza, Mínguez, Miguel, Remes-Troche, José-María, & Lacima, Glòria. Manometría anorrectal de alta resolución y de alta definición: redescubriendo la función anorrectal. Revista Española de Enfermedades Digestivas. 2018;110(12):794-805.