Pediatría
ISSN impreso:0120-4912
e-ISSN:2444-9369
DOI: 10.14295/rp.v56i2.424
Reporte de caso
Síndrome de Goldenhar, un abanico de posibilidades clínicas
Goldenhar syndrome, a range of clinical possibilities
Gloria Valentina Mosquera Sepúlvedaa, Mario Nicolas Ayala Lozanoa, Carlos Orlando Amaya Jinetea, Lorena García Agudeloa,b, José Luis Cruz Urregoa, Julio César Velasco Castroa,.
a. Médico. Hospital Regional de la Orinoquía, Yopal, Colombia.
b. Médica epidemióloga, Hospital Regional de la Orinoquía, Yopal, Colombia.
Recibido 13 de diciembre de 2022 Aceptado 06 de julio de 2023
Como Citar: Mosquera Sepúlveda GV. Ayala Lozano MN. Amaya Jinete CO. García Agudelo L. Cruz Urrego JL. Velasco Castro JC. Síndrome de
Goldenhar, un abanico de posibilidades clínicas. Pediatr. 2023;56(2): e424.
Autor para correspondencia: Gloria Valentina Mosquera Sepúlveda
Correo electrónico: valitamose@hotmail.es
Editor Jefe: Fernando Suárez-Obando
Resumen
Antecedentes: El síndrome de Goldenhar es una entidad clínica poco conocida, que ocasiona malformaciones principalmente en cara, ojos, columna vertebral y orejas. Afecta con mayor frecuencia al sexo masculino, el 85 % de los casos compromete la hemicara, siendo el lado derecho el más común. Reporte de caso: Recién nacido masculino, producto de primera gestación, madre de 19 años, sin antecedentes y adecuado control prenatal, al nacimiento evidenciaron múltiples malformaciones craneofaciales descritas como quiste dermoide escleral en ojo derecho que comprometía la región temporal inferior y superior de la córnea y un coloboma, pabellón auricular con microtia derecha y en hemicara ipsilateral lesión epidérmica levemente elevada, irregular, bordes definidos, con compromiso malar, cigomático y preauricular con micropápulas, además de lesión dermoide sugestiva de aplasia de cutis. Por todas las anomalías descritas fue diagnosticado con síndrome de Goldenhar. Conclusiones: El origen fisiopatológico no está bien definido, sin embargo, se ha descrito que es causado por una alteración de las estructuras derivadas del primer y segundo arco braquial, probablemente influenciado por factores externos pocos conocidos. En este caso, aunque la gestante asistió a todos los controles prenatales, esta alteración congénita solo se evidenció en el momento del parto, por lo que destacamos la pericia de los clínicos para reconocer las anomalías poco frecuentes y así, realizar un diagnóstico oportuno y un abordaje eficaz con un equipo multidisciplinario ya que condicionan un mejor pronóstico y calidad de vida en los pacientes.
Palabras clave: Malformaciones, síndrome de Goldenha, síndrome oculo-auriculo-vertebral, anomalías craneofaciales, recién nacido.
Abstract
Background: Goldenhar syndrome is a little-known clinical entity, that causes malformations mainly in the face, eyes, spine, and ears. It most frequently affects males; 85 % of the cases involve the hemiface, with the right side being the most common. Case report: Male newborn, product of first gestation, 19-year-old mother, without pathologic history and adequate prenatal control, at birth evidenced multiple craniofacial malformations described as a scleral dermoid cyst in the right eye involving the lower temporal and upper corneal regions and a coloboma, an auricular pavilion with right microtia, and in the ipsilateral hemiface a slightly elevated epidermal lesion, irregular, with defined borders, with malar, zygomatic, and preauricular involvement with micropapules, in addition to a dermoid lesion suggestive of skin aplasia. For all the anomalies described, he was diagnosed with Goldenhar syndrome. Conclusions: The pathophysiologic origin is not well defined; however, it has been described as being caused by an alteration of the structures derived from the first and second brachial arches, influenced by little-known external factors. In this case, although the pregnant woman attended all prenatal controls, this congenital alteration was only evidenced at the time of delivery, so we emphasize the expertise of clinicians to know the rare anomalies and thus make a timely diagnosis and an effective approach with a multidisciplinary team as they condition a better prognosis and quality of life in patients.
Key words: Malformations, goldenhar Syndrome, syndrome oculoauriculovertebral, craniofacial abnormalities, newborn
Introducción
El síndrome de Goldenhar (GHS) fue descrito por primera vez por Carl Ferdinand Von Arlt en 1845, posteriormente en 1962, Maurice Goldenhar, lo clasificó como entidad clínica independiente y más tarde, Cohen lo denominó displasia óculo auriculo vertebral (1). Se trata de una enfermedad congénita poco conocida que cursa con alteraciones de las estructuras derivadas del primer y segundo arco braquial, ocasionando malformaciones en cara, ojos, columna vertebral y orejas, teniendo esta última asociación a pérdida auditiva (1,2).
El GHS es el segundo tipo de malformación craneofacial más frecuente, su incidencia no está bien establecida se conoce que oscila entre 1/3500 o 1/15600 e incluso llegar a 1:45,000 casos/ nacidos vivos (2). Afecta mayormente al sexo masculino (relación hombre: mujer 3:2), el 85 % de los casos compromete la hemicara, siendo el lado derecho el más frecuente, mientras, el bilateral ocurre en 10-33 % (2,3).
La etiología de la enfermedad no está bien definida, sin embargo, se cree está influenciada con por múltiples factores externos como el consumo de medicamentos vasoactivos, tabaco, cocaína, talidomida, terapia hormonal y medicamentos antineoplásico, que alteran los procesos vasculares del mesoblasto hacia la cuarta semana del embarazo, afectando las estructuras craneofaciales derivadas del primer y segundo arco braquial (3).
En cuanto a las manifestaciones clínicas suelen ser diversas, pueden ser leves o complejas. Clínicamente, el GHS se caracteriza por la tríada clásica de microsomía craneofacial, quistes dermoides oculares y anomalías vertebrales (3,4).
El objetivo del reporte es presentar el caso de un recién nacido masculino sin antecedentes familiares que desarrolló GHS.
Reporte de caso
Recién nacido masculino, producto de primera gesta de madre de 19 años, aparentemente sana y adecuado control prenatal. El paciente nació por cesárea por bienestar fetal insatisfactorio y líquido amniótico con meconio grado II, llora y respira al nacer con APGAR 8-9-10, sin dificultad respiratoria, peso 3 750 gr, talla 50 cm y perímetro cefálico 35 cm.
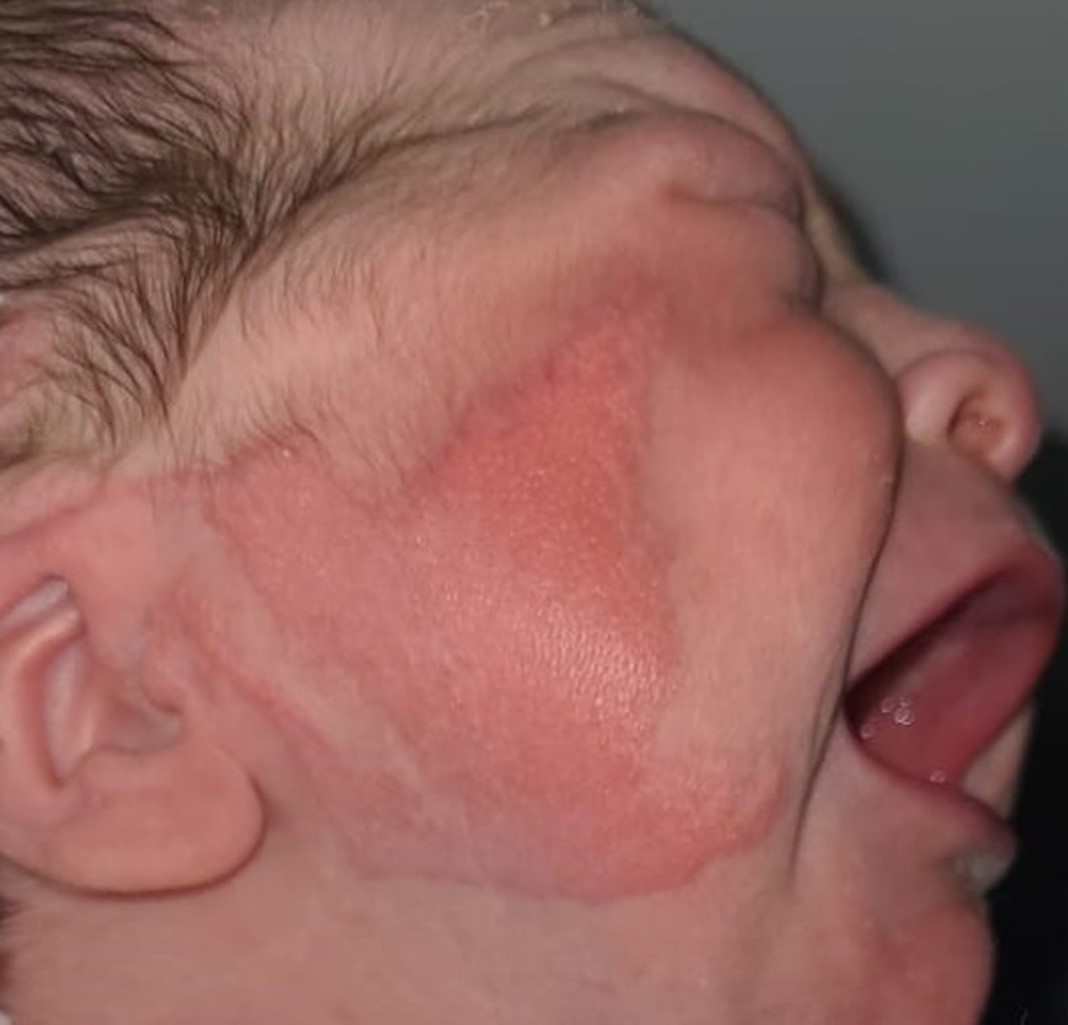
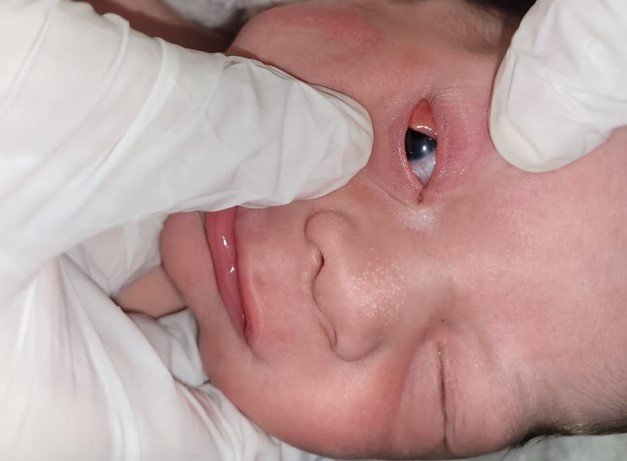
Al examen físico presentaba un quiste dermoide escleral en el ojo derecho que comprometía la región temporal inferior y superior de la córnea, sin causar limitación de la movilidad ocular (ver imagen 1), la fundoscopia con dilatación pupilar mostró vitreo claro, excavación de 0,1mm bordes regulares, emergencia central de vasos sanguíneos y mácula con brillo foveolar, con una fisura en el iris que se extendía desde la pupila hasta la comisura lateral de los párpados sugestiva de un coloboma, además de un área eritematosa que comprometía gran parte de la mucosa conjuntival (ver imagen 2). mientras, en hemicara derecha presentó lesión epidérmica levemente elevada, irregular, bordes definidos, con compromiso malar, cigomático y preauricular de 4x5 cm (ver imagen 3) con micropápulas. Ojo izquierdo sin alteraciones y pabellón auricular con microtia derecha.
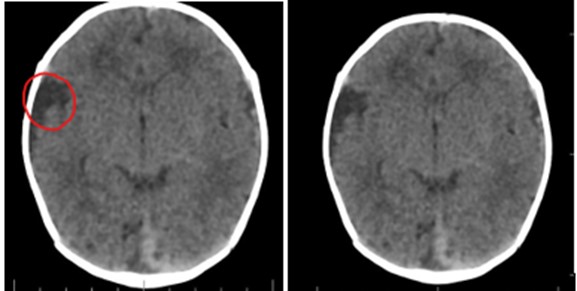
Por lesión dermoide consideraron que el paciente cursaba con aplasia cutis y en conjunto con las demás alteraciones estructurales del ojo y hemicara derecha (quiste dermoide escleral, coloboma y microtia) realizaron diagnóstico de síndrome de Goldenhar. Con la intención de descartar otras malformaciones asociadas, solicitaron tomografía axial computarizada (TAC) de cráneo que reportó quiste aracnoideo en la fosa temporal derecha y mega cisterna magna como variante anatómica (ver imagen 4). La TAC de columna cervical, ecocardiografía y ecografía de vías urinarias fueron normales.
Durante la hospitalización, el recién nacido (RN) desarrolló ictericia precoz (Kramer III e hiper bilirrubinemia; Bilirrubina total 13,39 mg/dL y B. directa 0.61 mg/dL) secundario a incompatibilidad de grupo sanguíneo (madre O positivo y RN A positivo) por lo que fue manejado con fototerapia simple durante dos días con resolución clínica completa.
El equipo interdisciplinario, consideró que el quiste dermoide escleral no ameritaba manejo quirúrgico de urgencia. Por evolución clínica favorable y hallazgos imagenológicos normales, dieron egreso, con manejo ambulatorio oftalmológico (carboximetilcelulosa 0,5 % gotas oftálmicas cada 6 horas y Polimixina B 0,1 % + Neomicina 0,35 % + dexametasona 6.000 UI/ mL en solución oftálmica cada 6 horas por 7 días) y ordenes de tamizaje auditivo y visual.
Figura 1. Quiste dermoide escleral
Figura 2. Mucosa conjuntival eritematosa que llega a comprometer la pupila.
Figura 3. Aplasia cutis.
Figura 4. Tomografía cráneo. Quiste aracnoideo en la fosa temporal derecha y mega cisterna magna como variante anatómica.
Discusión
El GHS es un trastorno polimalformativo, de origen esporádico cuya etiología y patogénesis no está bien establecida, sin embargo, González et al. (4) describen que puede asociarse con la administración de múltiples fármacos (talidomida, ácido retinoico, tamoxifeno), consumo de sustancias psicoactivas, enfermedades metabólicas (diabetes mellitus) e infecciosas (rubeola e influenza) durante la gestación. mientras, Zenteno et al. (5) afirman se han reportado casos hereditarios con patrones de herencia autosómico dominantes y recesivos que alteran el desarrollo y la expresión normal de las estructuras derivadas del primero y segundo arco faríngeo (3-5).
Clínicamente, el GHS se caracteriza por presentar disostosis craneofacial (6), en la que se observan anomalías oculares (65 %) de las cuales el 70 % son unilaterales; usualmente se presentan colobomas palpebrales que afectan el canto externo, párpados principalmente el superior y microftalmía. Con respecto al pabellón auricular, estas alteraciones son las más consistentes (6,7), Touliatou et al. (7) describen que pueden ocurrir en el 94 % de los pacientes, siendo la microtia unilateral e hipoacusia conductiva (76 %) las más frecuentes. Con respecto a la afectación neurológica los pacientes pueden desarrollar microcefalia, hidrocefalia, encefalocele occipital, malformación de Arnold Chiari, espina bífida y retraso mental en un 5-15 %.
En un reporte de caso informaron que los pacientes con GHS, pueden tener presentaciones clínicas atípicas con defectos oculares, retraso en el desarrollo y del habla, aunque las anomalías craneofaciales o vertebrales son típicas es importante tener en cuenta que pueden ocurrir casos inusuales con afectación neurológica predominante (8). En el caso presentado las anomalías físicas y/o estructurales fueron propias de la enfermedad, y dado la edad temprana de nuestro paciente no fue posible determinar manifestaciones neurológicas secundarias.
Entre otras alteraciones oculares puede versen tumores epibulbares, dermoides o lipodermoides, en el 35 % de los casos. Otros órganos que pueden estar comprometidos son: el corazón en un 5-58 %, con defectos septales interventriculares, ductus arterioso persistente, coartación aórtica, tetralogía de Fallot y transposición de los grandes vasos; musculoesqueléticos como la hipoplasia o aplasia del maxilar, temporal y malar, pie zambo, aplasia radial, entre otros y a nivel renal con ectopia, fusión y agenesia renal (9).
En el caso reportado, se sospechó GHS por el conjunto de características clínicas relacionadas, como aplasia cutis, quiste dermoide escleral, coloboma, microtia leve, además, del hallazgo imagenológico de quiste aracnoideo en la fosa temporal derecha y mega cisterna magna como variante anatómica aislada, algunos casos reportados en la literatura describen la presencia de quistes dermoides intracraneales en menor frecuencia de presentación (10).
Dado que el GHS tiene una heterogeneidad clínica, es importante descartar otras malformaciones asociadas (11). El neonato de este caso, ante la sospecha de un síndrome malformativo relacionado con GHS, descartaron anomalías vertebrales, urinarias y craneales, mediante estudios imagenológicos. Se resalta la presencia de hiperbilirrubinemia, sin embargo, por los estudios se determinó que se presentó por incompatibilidad de grupo.
El presente caso clínico reúne 2 de los criterios de M. Feingold y J. Murray consistentes en microtia y coloboma, sin embargo, hay otras alteraciones que presentó el neonato, no indicadas en la clasificación sugerida como deformidades en iris, quiste dermoide escleral, aplasia cutis, que también son características de la enfermedad (12), por tal razón la sospecha clínica no debe limitarse a las alteraciones físicas ya que pueden presentarse alteraciones menos evidentes y de menor gravedad que definen el síndrome y que no descartan su diagnóstico (12,13).
El pronóstico de los pacientes con GHS depende en gran medida de las diferentes alteraciones anatómicas y funcionales que presenten. En los casos atípicos donde las manifestaciones oftalmológicas, auditivas y neurológicas no son tan evidentes por lo que el seguimiento con un equipo multidisciplinario-especializado permitirá identificar estas manifestaciones pocos frecuentes y así, brindar manejos oportunos que mejoren la calidad de vida de estos pacientes (14).
Conclusiones
El origen fisiopatológico del GHS no está bien dilucidado, sin embargo, se ha descrito que es causada por una alteración de las estructuras derivadas del primer y segundo arco braquial, probablemente influenciado de factores externos pocos conocidos. En este caso, aunque la gestante asistió a todos los controles prenatales, esta alteración congénita solo se evidenció en el momento del parto, por lo que destacamos la pericia de los clínicos para reconocer las anomalías poco frecuentes y así, realizar un diagnóstico oportuno y un abordaje eficaz con un equipo multidisciplinario ya que condicionan un mejor pronóstico y calidad de vida en los pacientes.
Agradecimientos
Al Hospital Regional de la Orinoquia.
Conflicto de intereses
Ninguno declarado por los autores.
Financiación
No se recibió ningún tipo de financiación.
Referencias
1. Agredo FE, Cuello G, Blanco P. Síndrome de Golden Har. Reporte de un caso. Acta otorrinolaringol cir cabeza cuello [Internet]. 31 de agosto de 2018;37(4):215-9. Disponible en: https://revista.acorl.org.co/index.php/acorl/article/view/302.
2. Guevara-Valmaña OI, Nahas-Combina L, Andrade-Delgado L, Apellaniz-Campo AG, Leyva-Sotelo LM, Gaspar-Daniel Á. Goldenhar syndrome: Surgical management protocol in a reference center. Cir y Cir (English Ed. 2019;87(5):516–27.
3. Torres Salinas C. Síndrome de Goldenhar: Manifestaciones clínicas y revisión de literatura. Revista pediátrica de Panamá- 2020, Marzo, 49(1):17-20. doi: 10.37980/im.jour- nal.rspp2019.1591.
https://doi.org/10.37980/im.journal.rspp2019.1591.4. González L, Ramos A, Lozano S, Salazar R, López C. Síndrome de Goldenhar: a propósito de un caso. Rev Pediatr Aten Primaria. 2016 Mar; 18(69): 49-53.
5. Zenteno-Salazar E, Sifuentes-Vela C, Contreras-Capetillo S, López-Cabrera M, Núñez-Enríquez JC. Multidisciplinary management with neuromuscular electrical stimulation therapy in a Goldenhar syndrome patient who presented swallowing disorder and failure to thrive. Bol Med Hosp Infant Mex. 2021;78(4):362-369. Spanish. doi: https://doi.org/10.24875/BMHIM.20000222.
6. Guerrero-Domínguez R, López-Herrera-Rodríguez D, Benítez-Linero I, Ontanilla A. Manejo anestésico para cirurgia de atresia de esôfago em um recém-nascido com síndrome de Goldenhar [Anesthetic management for surgery of esophagus atresia in a newborn with Goldenhar’s syndrome]. Rev Bras Anestesiol. 2015 Jul-Aug;65(4):298-301. Portuguese. doi: https://doi.org/10.1016/j.bjan.2013.07.011.
7. Touliatou V, Fryssira H, Mavrou A, Kanavakis E, Kitsiou-Tzeli S. Clinical manifestations in 17 Greek patients with Goldenhar syndrome. Genet Couns. 2006;17(3):359-70.
8. Jayaprakasan SK, Waheed MD, Batool S, Pimentel Campillo J, Nageye ME, Holder SS. Goldenhar Syndrome: An Atypical Presentation With Developmental and Speech Delay. Cureus. 2023 Mar 16;15(3):e36225. doi: 10.7759/cureus.36225.
9. El Bouaychi A, Ez-Zahraoui M, Boutimzine N, Cherkaoui O. Syndrome de Goldenhar: à propos d’un cas [Goldenhar syndrome: A case report]. J Fr Ophtalmol. 2020 Jan;43(1):90-92. French. doi: https://doi.org/10.1016/j.jfo.2019.07.006.
10. Barisic I, Odak L, Loane M, Garne E, Wellesley D, Calzolari E, Dolk H, Addor MC, Arriola L, Bergman J, Bianca S, Doray B, Khoshnood B, Klungsoyr K, McDonnell B, Pierini A, Rankin J, Rissmann A, Rounding C, Queisser-Luft A, Scarano G, Tucker D. Prevalence, prenatal diagnosis and clinical features of oculo-auriculo-vertebral spectrum: a registry-based study in Europe. Eur J Hum Genet. 2014 Aug;22(8):1026-33. doi: https://doi.org/10.1038/ejhg.2013.287.
11. Feingold M, Baum J. Goldenhar´s Syndrome. Am J Dis Child 1978;132(2):136-138.doi: https://doi.org/10.1001/archpedi.1978.0212027%200034006.
12. Tam MW, Boyle N. Goldenhar syndrome associated with lacrimal system agenesis: A case report. Am J Ophthalmol Case Rep. 2022 Dec 6;29:101766. doi: https://doi.org/10.1016/j.ajoc.2022.101766.
13. Liao EN, Chan DK. Congenital aural atresia and first branchial cleft anomalies: Cholesteatoma and surgical management. Laryngoscope Investig Otolaryngol. 2022 Apr 20;7(3):863-869. doi: https://doi.org/10.1002/lio2.793.
14. Malta PG, Vilani DSRA, de Miranda CF, Fouad IL, Castro CC, Borgatti MÉ. Goldenhar syndrome: the importance of an ophthalmological approach. Rom J Ophthalmol. 2020 Oct-Dec;64(4):444-448. https://doi.org/10.22336/rjo.2020.68.