
Mucopolysaccharidosis type I, Hurler syndrome variant : Initial approach and relationship to literature
Article Sidebar
Main Article Content
Abstract
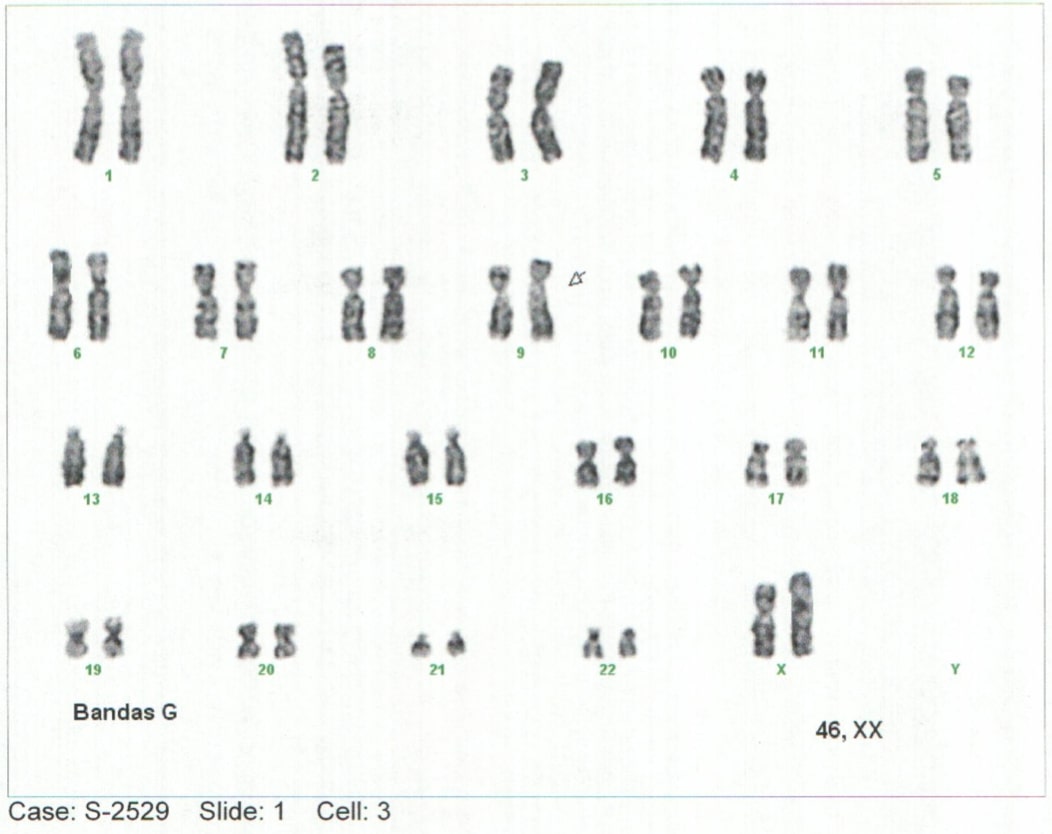
Background: Mucopolysaccharidoses-type storage diseases are a group of rare genetic diseases with an autosomal recessive inheritance pattern. Mucopolysaccharidosis (MPS) is a condition of lysosomal overload caused by deficiencies of enzymes responsible for the degradation of glycosaminoglycans (GAG), also called mucopolysaccharides. This enzyme deficiency is generated from the progressive accumulation of compounds in different tissues that leads to generalized tissue damage and tends to progress to multiorgan failure (1–5). Case report: Elderly lactating female patient with neurodevelopmental delay and notable phenotypic alterations, which is related to findings described in the literature. Conclusions: alpha-L-Iduronidase enzyme deficiency was identified in the context of a clinical picture with severe manifestations and such an early age of onset of the pathology, it is classified as MPS I, or Hurler Syndrome.Advances in the early approach and knowledge of the natural history of deposit diseases will make it possible to generate a better diagnostic and therapeutic approach, generating a better outcome.
Downloads
Article Details

This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
Creative Commons
License Attribution-NonCommercial-ShareAlike 4.0 International (CC BY-NC-SA 4.0)
You are free to:
Share - copy and redistribute the material in any medium or format.
Adapt - remix, transform, and build upon the material The licensor cannot revoke these freedoms as long as you follow the license terms.
• Attribution — You must give appropriate credit, provide a link to the license, and indicate if changes were made. You may do so in any reasonable manner, but not in any way that suggests the licensor endorses you or your use.
• NonCommercial — You may not use the material for commercial purposes.
• ShareAlike — If you remix, transform, or build upon the material, you must distribute your contributions under the same license as the original.
• No additional restrictions — You may not apply legal terms or technological measures that legally restrict others from doing anything the license permits.




References
Moore D, Connock MJ, Wraith E, Lavery C. The prevalence of and survival in Mucopolysaccharidosis I: Hurler, Hurler-Scheie and Scheie syndromes in the UK. Orphanet J Rare Dis. 2008;3(1):1–7. DOI: https://doi.org/10.1186/1750-1172-3-24
Filocamo M, Tomanin R, Bertola F, Morrone A. Biochemical and molecular analysis in mucopolysaccharidoses: what a paediatrician must know. Ital J Pediatr. 2018;44(Suppl 2):129. DOI: https://doi.org/10.1186/s13052-018-0553-2
Feillet F, Wiedemann A, Jeannesson E, Jaussaud R, Journeau P. Mucopolisacaridosis. EMC - Pediatría. 2016;51(3):1–14. DOI: https://doi.org/10.1016/S1245-1789(16)78912-8
Tebani A, Zanoutene-Cheriet L, Adjtoutah Z, Abily-Donval L, Brasse-Lagnel C, Laquerrière A, et al. Clinical and molecular characterization of patients with mucopolysaccharidosis type I in an Algerian series. Int J Mol Sci. 2016;17(5). DOI: https://doi.org/10.3390/ijms17050743
Khan SA, Peracha H, Ballhausen D, Wiesbauer A, Gautschi M, Mason RW, et al. Molecular Genetics and Metabolism. HHS Public Access Author. 2018;121(3):227–40. DOI: https://doi.org/10.1016/j.ymgme.2017.05.016
Spranger WJ, Kliegman R. Nelson. Tratado de pediatria - Mucopolisacaridosis [Internet]. 20th Editi. Nelson. Tratado de pediatría. Elsevier Espa8#241;a, S.L.U.; 2016. 772–779 p. Available from: https://www-clinicalkey-es.ez.unisabana.edu.co/#!/browse/book/3-s2.0-C20151024243
Simon Jones, MD, Robert Wynn M. Clinical features and diagnosis. UpToDate [Internet]. 2019; Available from: https://www-uptodate-com.ez.unisabana.edu.co/contents/mucopolysaccharidoses-clinical-features-and-diagnosis/print?search=mucopolisacaridosis&source=search_…
Lee-Chen GJ, Wang TR. Mucopolysaccharidosis type I: Identification of novel mutations that cause Hurler/Scheie syndrome in Chinese families. J Med Genet. 1997;34(11):939–41. DOI: https://doi.org/10.1136/jmg.34.11.939
Yang C, Pan J, Linpeng S, Li Z, Tan H, Wu L. Identification of five novel mutations causing rare lysosomal storage diseases. Med Sci Monit. 2019;25:7634–44. DOI: https://doi.org/10.12659/MSM.915876
Scott HS, Bunge S, Gal A, Clarke LA, Phillip Morris C, Hopwood JJ, et al. Molecular Genetics of Mucopolysaccharidosis Type I: Diagnostic, Clinical, and Biological Implications. Hum Mutat. 1995;6:288302. DOI: https://doi.org/10.1002/humu.1380060403
Bianchi PM, Gaini R, Vitale S. ENT and mucopolysaccharidoses. Ital J Pediatr. 2018;44(Suppl 2):127. DOI: https://doi.org/10.1186/s13052-018-0555-0
Suarez-Guerrero JL, Gómez Higuera PJI, Arias Flórez JS, Contreras-García GA. Mucopolisacaridosis: características clínicas, diagnóstico y de manejo. Rev Chil Pediatr. 2016;87(4):295–304. DOI: https://doi.org/10.1016/j.rchipe.2015.10.004
Clarke LA, Atherton AM, Burton BK, Day-Salvatore DL, Kaplan P, Leslie ND, et al. Mucopolysaccharidosis Type I Newborn Screening: Best Practices for Diagnosis and Management. J Pediatr [Internet]. 2017;182:363–70. Available from: http://dx.doi.org/10.1016/j.jpeds.2016.11.036 DOI: https://doi.org/10.1016/j.jpeds.2016.11.036
Kiely BT, Kohler JL, Coletti HY, Poe MD, Escolar ML. Early disease progression of Hurler syndrome. Orphanet J Rare Dis. 2017;12(1):1–10. DOI: https://doi.org/10.1186/s13023-017-0583-7
Harmatz PR, Shediac R. Mucopolysaccharidosis VI: Pathophysiology, diagnosis and treatment. Front Biosci - Landmark. 2017;22(3):385–406. DOI: https://doi.org/10.2741/4490
Coutinho MF, Encarnação M, Matos L, Silva L, Ribeiro D, Santos JI, et al. Molecular characterization of a novel splicing mutation underlying mucopolysaccharidosis (MPS) type VI—Indirect proof of principle on its pathogenicity. Diagnostics. 2020;10(2):1–11. DOI: https://doi.org/10.3390/diagnostics10020058
Peck DS, Lacey JM, White AL, Pino G, Studinski AL, Fisher R, et al. Incorporation of second-tier biomarker testing improves the specificity of newborn screening for mucopolysaccharidosis type i. Int J Neonatal Screen. 2020;6(1):1–10. DOI: https://doi.org/10.3390/ijns6010010
Shafaat M, Hashemi M, Majd A, Abiri M, Zeinali S. Genetic testing of Mucopolysaccharidoses disease using multiplex PCR- based panels of STR markers: in silico analysis of novel mutations. Metab Brain Dis. 2019;34(5):1447–55. DOI: https://doi.org/10.1007/s11011-019-00434-z
Li X, Xiao R, Chen B, Yang G, Zhang X, Fu Z, et al. A novel mutation of SGSH and clinical features analysis of mucopolysaccharidosis type IIIA. Med (United States). 2018;97(52). DOI: https://doi.org/10.1097/MD.0000000000013758
Parini R, Deodato F, Di Rocco M, Lanino E, Locatelli F, Messina C, et al. Open issues in Mucopolysaccharidosis type I-Hurler. Orphanet J Rare Dis. 2017;12(1):1–9. DOI: https://doi.org/10.1186/s13023-017-0662-9
Sawamoto K, Chen HH, Alméciga-Díaz CJ, Mason RW, Tomatsu S. Gene therapy for Mucopolysaccharidoses. Mol Genet Metab. 2018;123(2):59–68. DOI: https://doi.org/10.1016/j.ymgme.2017.12.434
Jameson E, Jones S, Remmington T. Enzyme replacement therapy with laronidase (Aldurazyme®) for treating mucopolysaccharidosis type I. Cochrane Database Syst Rev. 2019;2019(6). DOI: https://doi.org/10.1002/14651858.CD009354.pub5
Fecarotta S, Gasperini S, Parenti G. New treatments for the mucopolysaccharidoses: from pathophysiology to therapy. Ital J Pediatr. 2018;44(Suppl 2):124. DOI: https://doi.org/10.1186/s13052-018-0564-z
Kuiper GA, Nijmeijer SCM, Roelofs MJM, van der Lee JH, Hollak CEM, Bosch AM. Limited data to evaluate real-world effectiveness of enzyme replacement therapy for mucopolysaccharidosis type I. J Inherit Metab Dis. 2019;42(5):762–75. DOI: https://doi.org/10.1002/jimd.12103
Eisengart JB, Rudser KD, Xue Y, Orchard P, Miller W, Lund T, et al. Long-term outcomes of systemic therapies for Hurler syndrome: an international multicenter comparison. Genet Med. 2018;20(11):1423–9. DOI: https://doi.org/10.1038/gim.2018.29